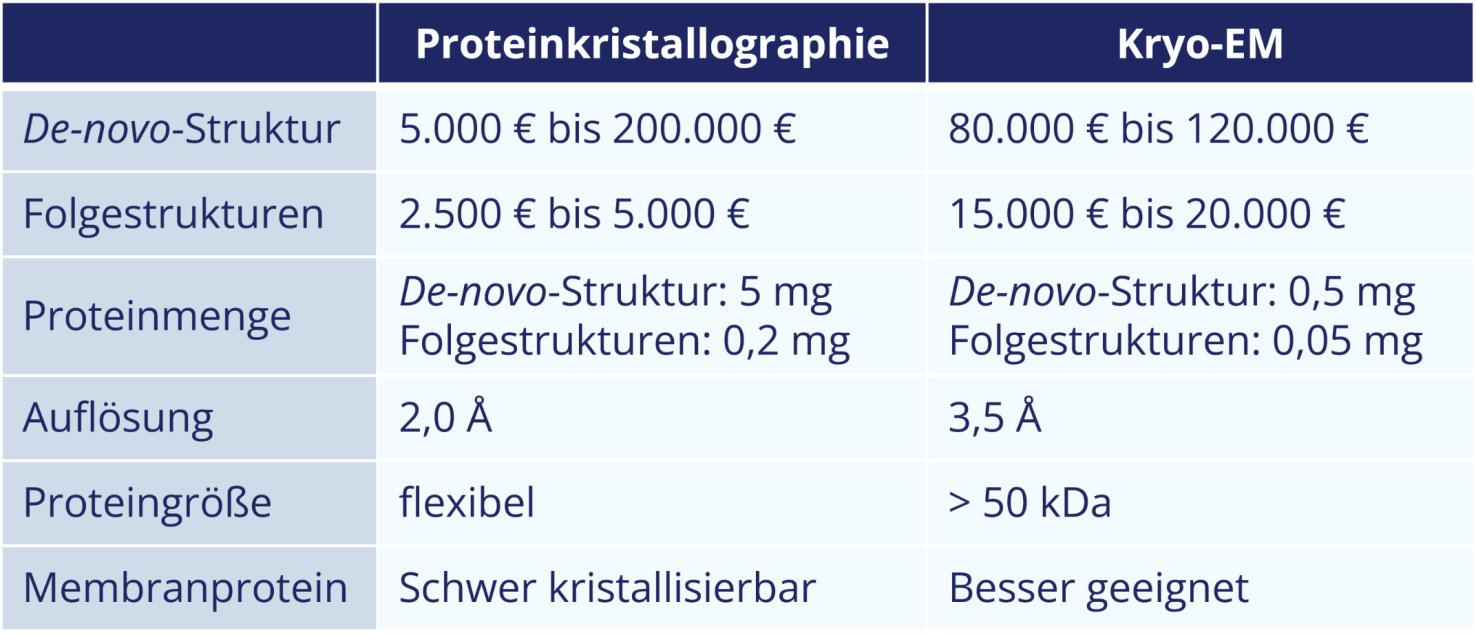
Die proteinkristallographische Bestimmung einer De-novo-Struktur beginnt bei etwa 5.000 € und kann durchaus 200.000 € betragen. Dies ist im Wesentlichen davon abhängig, wie hoch der Aufwand zur Optimierung der Kristallisationsbedingung ist und wie leicht sich die Phasen sowie die Kryo-Bedingung bestimmen lassen.
Die Folgestrukturen liegen in der Regel im Bereich von 2.500 € bis 5.000 €. Dies hängt davon ab, wieviel Erfahrung man mit dem Protein hat, wie gut die Modellstruktur ist, ob ein Skript für die Strukturlösung verwendet wird, welchen Verfeinerungsstatus man benötigt und wie groß das Protein ist (Aminosäuren, Moleküle in der asymmetrischen Einheit).
Die De-novo-Strukturbestimmung mittels Kryo-EM ist deutlich besser zu prognostizieren. Hier sollte man mit etwa 100.000 € rechnen. Der größte Aufwand liegt in der Bestimmung der richtigen Bedingungen für die Herstellung der Grids. Für die Folgestrukturen sollte man mit etwa 15.000 € bis 20.000 € kalkulieren.
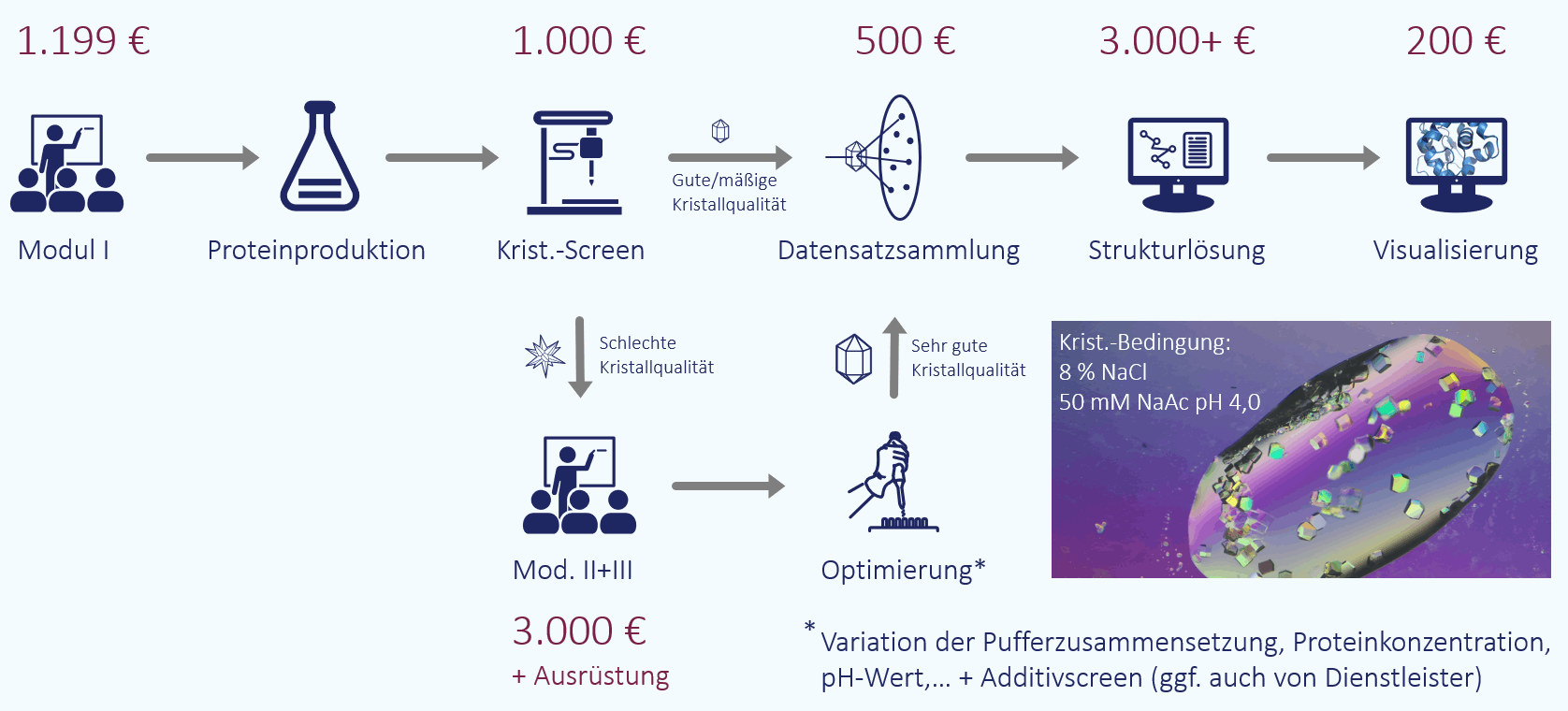
Aufgrund der niedrigeren Kosten, ist es empfehlenswert, mit der proteinkristallographischen Strukturbestimmung zu beginnen, d.h. man gibt einen Kristallisationsscreen in Auftrag und schaut, ob das Protein kristallisiert. Im Idealfall, lässt sich der Kristall direkt am Synchrotron vermessen und die Struktur lösen. Hier sind 5.000 € durchaus realistisch, obwohl man besser mit 10.000 € bis 15.000 € kalkuliert, insbesondere, wenn die Struktur in der Proteindatenbank deponiert werden soll.
Kryo-EM ist zu empfehlen, wenn man die Struktur eines Membranproteins bestimmen möchte, einem sehr wenig Protein zur Verfügung steht oder im Rahmen eines Kristallisationsscreenings keine geeignete Kristallisationsbedingung gefunden wurde. Allerdings hat die Kryo-EM, neben den höheren Kosten, den Nachteil, dass die Auflösung meistens deutlich schlechter ist, so dass man keine klare Aussage bezüglich der Ausrichtung einer Seitenkette oder des Inhibitors treffen kann. Zudem ist die Struktur von Proteinen mit einem Molekulargewicht kleiner 50 kDa mittels Kryo-EM nicht bestimmbar. Die Fusionierung mit einem weiteren Protein kann hier eventuell zum Erfolg führen.