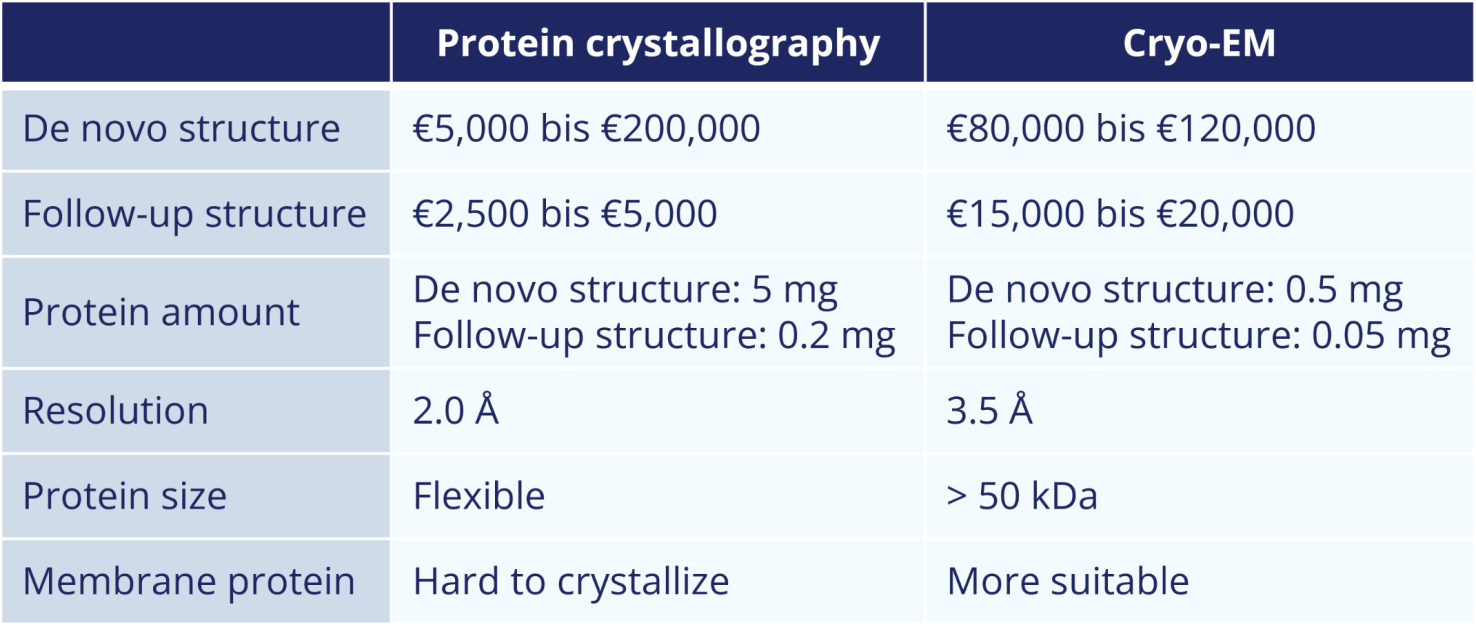
The protein crystallographic determination of a de novo structure starts at around €5,000 and can easily amount to €200,000. This essentially depends on how much effort is required to optimize the crystallization condition and how easily the phases and the cryo condition can be determined.
The subsequent structures are usually in the range of €2,500 to €5,000. This depends on how much experience you have with the protein, how good the model structure is, whether a script is used for the structure solution, what refinement state you need, and the size of the protein (amino acids and molecules in the asymmetric unit).
The de novo structure determination using cryo-EM is much better for predicting the costs. Expect to pay around €100,000 here. The greatest effort lies in determining the right conditions for the production of the grids. For the subsequent structures you should calculate with about 15,000 € to 20,000 €.
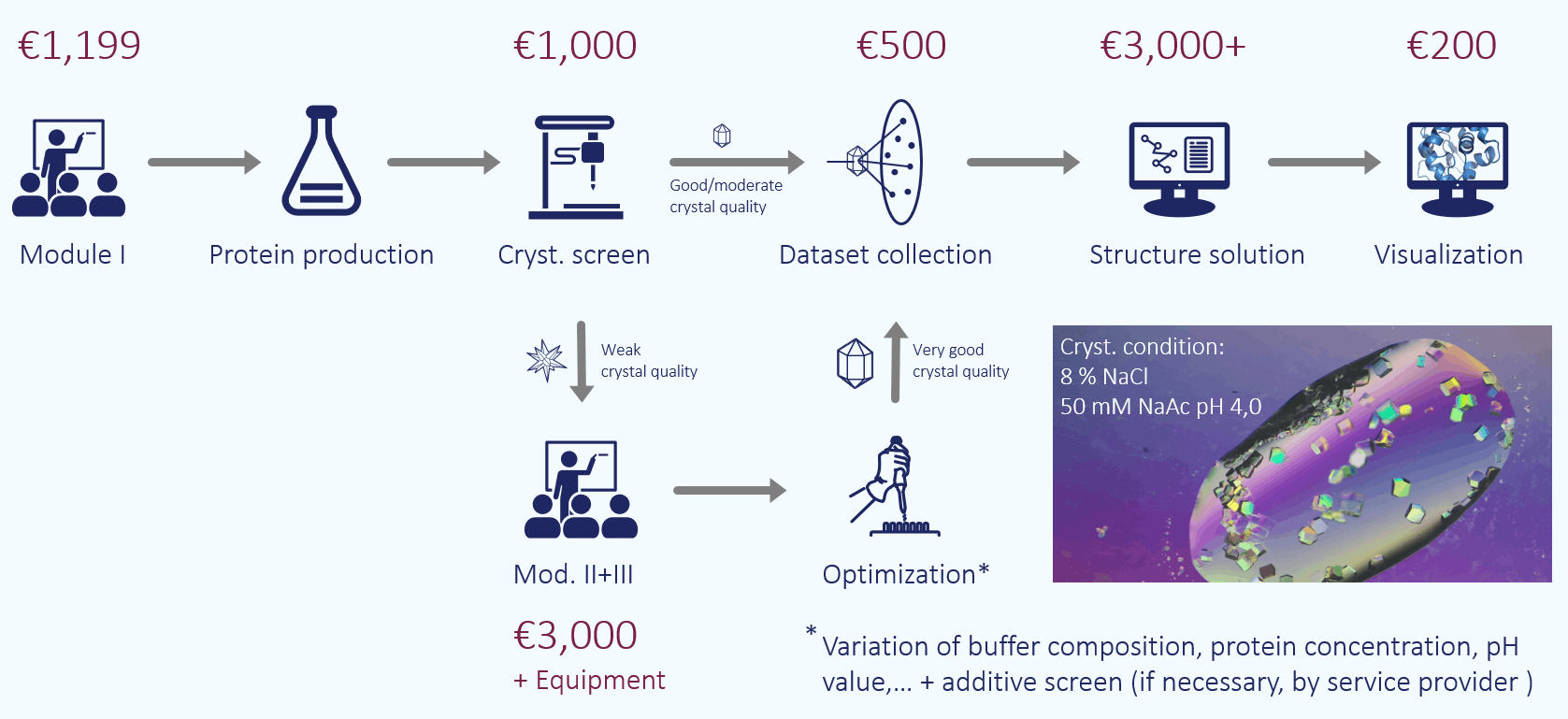
Due to the lower costs, it is recommended to start with protein crystallographic structure determination, i.e. commission a crystallization screen and see if the protein crystallizes. Ideally, the crystal can be measured directly at the synchrotron and the structure can be solved. €5,000 is quite realistic here, although it is better to calculate with €10,000 to €15,000, especially if the structure is to be deposited in the protein database.
Cryo-EM is recommended if you want to determine the structure of a membrane protein, if there is only a small amount of protein available, or if no suitable crystallization condition was found in a crystallization screening. However, in addition to the higher costs, cryo-EM has the disadvantage that the resolution is usually much lower, so that no clear statement can be made regarding the orientation of a side chain or the inhibitor. In addition, the structure of proteins with a molecular weight of less than 50 kDa cannot be determined using cryo-EM. Fusing with another protein can possibly lead to success here.